Summary
Wilson’s disease, also known as hepatolenticular degeneration and progressive lenticular degeneration, is a rare genetic disorder that primarily affects the liver, brain, and eyes. In this condition, excessive amounts of copper accumulate in these organs because the liver cannot properly filter out excess copper from the blood.
An individual may develop Wilson’s disease if they inherit a defective gene from both parents. Specifically, mutations in the ATP7B gene impair the liver’s ability to eliminate excess copper. As the disease progresses, the accumulated copper in the body can cause a golden-brown discoloration of the corneas called Kayser-Fleischer rings. It may also lead to yellowing of the skin and whites of the eyes (jaundice), abdominal pain and swelling, uncontrolled movements, and various other symptoms.
To treat this disease, doctors may prescribe medications such as chelating agents (e.g., penicillamine or trientine) that bind to copper and help remove it from the body, or zinc therapy to reduce copper absorption in the intestines. In severe cases, a liver transplant might be necessary to restore normal liver function and copper metabolism. Additionally, patients may be advised to follow a special low-copper diet and undergo physical therapy to manage neurological symptoms.
Table of Contents
Symptoms of Wilson’s Disease

Wilson’s disease presents a variety of symptoms that depend on the parts of the body affected by copper accumulation. The most commonly impacted areas are the liver, brain, and eyes. Recognizing these symptoms early is crucial for timely intervention.
Liver-Related Symptoms
When the liver is affected, the patient may experience:
- General Weakness. A persistent feeling of overall body fatigue and lack of energy.
- Frequent Fatigue. Constant tiredness that doesn’t improve with rest.
- Weight Loss. Unintentional loss of body weight due to decreased appetite.
- Nausea and Vomiting. Feelings of sickness accompanied by episodes of vomiting.
- Loss of Appetite. Reduced desire to eat, leading to nutritional deficiencies.
- Itching of the Skin. Persistent itchiness caused by the buildup of bile salts.
- Yellowing of the Skin and Eyes (Jaundice). A yellow tint due to excess bilirubin in the bloodstream.
- Swelling of the Abdomen and Legs. Fluid retention causing abdominal bloating and leg edema.
- Abdominal Pain and Bloating. Discomfort from liver enlargement or fluid accumulation.
- Visible Blood Vessels on the Skin. Spider-like veins appearing under the skin surface.
- Muscle Cramps. Sudden, involuntary contractions of muscles.
Neurological Symptoms
If the brain is affected, the patient may experience:
- Memory Difficulties. Trouble recalling information or recent events.
- Speech Problems. Difficulty articulating words or experiencing slurred speech.
- Vision Issues. Blurred or double vision and other visual disturbances.
- Difficulty Walking. Impaired coordination leading to unsteady gait or balance problems.
- Migraines. Severe headaches often accompanied by nausea or sensitivity to light.
- Excessive Salivation. Increased production of saliva causing drooling.
- Insomnia. Difficulty falling or staying asleep.
- Frequently Dropping Objects. Inability to hold onto items due to tremors or poor muscle control.
- Mood and Behavioral Changes. Irritability, mood swings, or changes in personality and behavior.
- Depression. Persistent feelings of sadness, hopelessness, and loss of interest in activities.
Eye Symptoms
Notable symptoms affecting the eyes include:
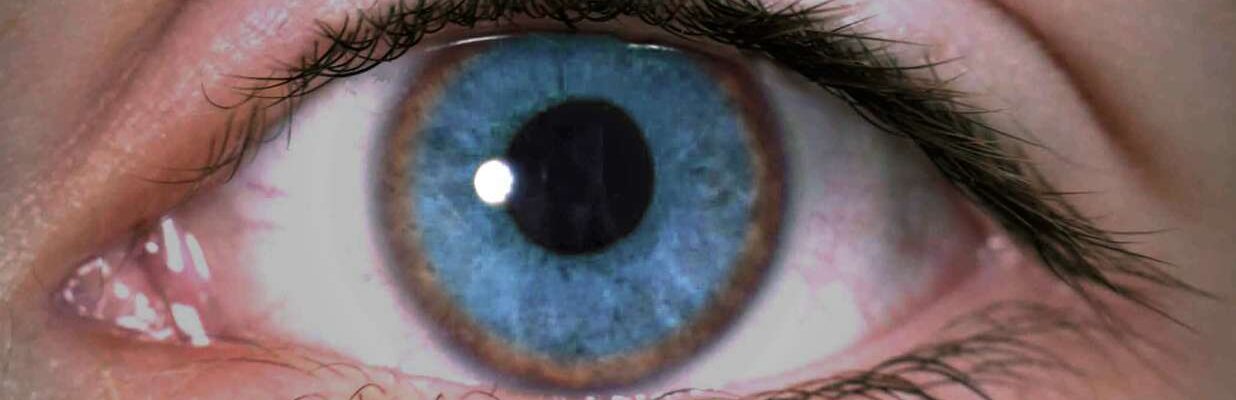
- Golden-Brown Discoloration of the Corneas (Kayser-Fleischer Rings). Copper deposits forming rings around the corneas, giving a golden-brown hue.
- Sunflower Cataracts. Multicolored, sunflower-like appearances in the lens of the eyes due to copper accumulation.
These symptoms often resemble those of other liver and neurological conditions, making Wilson’s disease challenging to identify based solely on clinical signs. However, the presence of Kayser-Fleischer rings and sunflower cataracts in the eyes are distinctive indicators of this condition. Recognizing these specific eye symptoms can be crucial for early detection and management.
If you or someone you know is experiencing these symptoms, especially in combination, it’s important to seek medical attention promptly. Early diagnosis and treatment can significantly improve outcomes and quality of life for individuals with Wilson’s disease.
Diagnostic Procedures for Wilson’s Disease
Diagnosing Wilson’s disease can be challenging because its symptoms often mimic those of other liver and neurological disorders. Early and accurate diagnosis is essential for effective treatment and prevention of serious complications. The following diagnostic procedures are commonly used to confirm the presence of Wilson’s disease:
- Blood Tests. These tests measure levels of copper, ceruloplasmin (a protein that binds copper in the blood), and liver enzymes. Low ceruloplasmin levels and abnormal liver enzyme levels may indicate Wilson’s disease.
- 24-Hour Urine Copper Test. This test assesses the amount of copper excreted in the urine over a 24-hour period. Elevated copper levels suggest improper copper metabolism and accumulation in the body.
- Eye Examination. An ophthalmologist conducts a slit-lamp examination to detect Kayser-Fleischer rings—golden-brown rings around the corneas caused by copper deposits—which are a key indicator of Wilson’s disease.
- Liver Biopsy. A small sample of liver tissue is extracted and analyzed for copper content. High levels of copper in the liver confirm the diagnosis and help assess the extent of liver damage.
- Genetic Testing. This involves analyzing the ATP7B gene for mutations responsible for Wilson’s disease. Identifying these mutations can confirm the diagnosis and is also useful for screening family members.
- Magnetic Resonance Imaging (MRI) and Computed Tomography (CT) Scans. Imaging studies of the brain and liver help detect abnormalities and assess the extent of organ damage caused by copper accumulation.
These diagnostic procedures enable healthcare professionals to accurately identify Wilson’s disease and differentiate it from other conditions with similar symptoms. Early detection allows for prompt treatment, which is crucial for preventing irreversible organ damage and improving the patient’s quality of life.
Complications of Untreated Wilson’s Disease
If Wilson’s disease is left untreated, the accumulation of copper in vital organs can lead to severe and potentially life-threatening complications. Early detection and management are crucial to prevent irreversible damage.
- Liver Failure. Excessive copper buildup causes chronic liver damage, leading to scarring (cirrhosis) and eventually liver failure, which impairs the liver’s ability to perform essential functions.
- Neurological Disorders. Copper accumulation in the brain can result in movement disorders such as tremors, muscle stiffness, difficulty with coordination, and speech problems, which may become permanent.
- Psychiatric Issues. Untreated copper toxicity can lead to depression, mood swings, irritability, and even psychosis, significantly affecting mental health and quality of life.
- Hemolytic Anemia. High copper levels can damage red blood cells, causing them to break down prematurely and leading to anemia characterized by fatigue and weakness.
- Kidney Damage. Copper deposition can impair kidney function, resulting in proteinuria (excess protein in urine) and decreased ability to filter waste from the blood.
- Bone and Joint Problems. Copper accumulation may cause osteoporosis and arthritis, leading to bone pain, fractures, and joint stiffness.
- Cardiac Issues. Copper can deposit in heart tissues, leading to arrhythmias (irregular heartbeats) and cardiomyopathy (disease of the heart muscle), increasing the risk of heart failure.
- Pancreatitis. Inflammation of the pancreas can occur, causing severe abdominal pain, nausea, and digestive problems.
- Death. Severe organ failure resulting from untreated Wilson’s disease can be fatal if not promptly addressed.
Untreated Wilson’s disease poses serious health risks, but with early diagnosis and appropriate treatment, many of these complications can be prevented or managed effectively. It is essential to seek medical attention if symptoms suggestive of Wilson’s disease are present to ensure timely intervention and improve the chances of a positive outcome.
Causes of Wilson’s Disease
The body requires an adequate amount of copper to keep nerves and skin healthy. Copper is typically obtained from the foods we eat and is eliminated from the body with the help of the liver. However, in individuals with Wilson’s disease, copper accumulates inside the body instead of being properly excreted.
The primary cause of Wilson’s disease is the inheritance of an abnormal or defective gene from one’s parents. This genetic mutation affects the liver’s ability to filter out excess copper from the bloodstream. As a result, the liver cannot effectively remove copper, leading to its buildup in the liver, brain, and eyes.
When this defective gene is inherited, the liver struggles to process and eliminate copper, causing it to accumulate to toxic levels. Over time, this excess copper can cause significant damage to vital organs, leading to the various symptoms associated with Wilson’s disease.
Understanding that Wilson’s disease is a genetic disorder highlights the importance of family medical history and genetic counseling. Early detection and intervention are crucial to prevent severe complications resulting from copper accumulation in the body.
Prevention of Wilson’s Disease

While Wilson’s disease cannot be entirely prevented since it is an inherited condition, the early onset and progression of the disease can be managed through the following measures:
- Avoid Foods High in Copper. To prevent excessive copper accumulation in the body, avoid consuming foods rich in copper such as chocolate, liver, mushrooms, nuts, and all types of shellfish (shrimp, prawns, crabs, mussels, oysters). Limiting these foods can help reduce copper intake.
- Install Water Filters on Faucets. Copper pipes can leach copper into the water supply. Using water filters can help remove excess copper from your drinking water, reducing overall copper consumption.
- Reduce or Avoid Alcohol Consumption. To maintain liver health, limit excessive alcohol intake. Alcohol contains substances that can damage liver cells, impairing the liver’s ability to filter out excess copper.
- Quit Smoking. Cigarettes contain toxic substances that can harm liver cells. A damaged liver may struggle to eliminate excess copper from the body effectively.
- Choose Nutritious Foods. Maintain the health of vital organs like the liver, brain, and eyes by consuming a balanced diet rich in essential nutrients. Eating wholesome foods supports overall body function and resilience.
- Exercise Daily. Regular physical activity helps eliminate toxins from the body through sweat. Exercise also helps maintain a healthy weight, reducing the risk of fatty liver disease, which can further compromise liver function.
- Drink Plenty of Water. Staying well-hydrated promotes regular urination, which aids in flushing out excess copper from the body. Adequate water intake supports kidney function and overall detoxification processes.
In addition to these measures, it is important to consult a doctor regularly to detect any health issues early on. Regular medical check-ups enable early diagnosis and prompt treatment of any conditions, including Wilson’s disease, thereby improving the chances of effective management and better health outcomes.
Risk Factors for Wilson’s Disease

Wilson’s disease is a genetic disorder, which means that individuals with a family history of the condition have the highest likelihood of developing it. Understanding these risk factors is crucial for early detection and management.
- Family History of Wilson’s Disease. Those who have relatives affected by Wilson’s disease are at a greater risk because the condition is inherited. If both parents are carriers of the abnormal gene, their children have a 25% chance of inheriting the disease.
- Carrier Parents. When both parents carry the defective gene but do not show symptoms themselves, they can still pass the gene to their offspring. Each child has a one in four chance of receiving two copies of the abnormal gene, leading to the development of Wilson’s disease.
- Age Range Between 5 and 35 Years. The symptoms of Wilson’s disease usually manifest between the ages of 5 and 35 years old. Being within this age group increases the likelihood of symptom onset, although the disease can occasionally present earlier or later.
It’s important for individuals with a known family history of Wilson’s disease to consider genetic counseling and undergo regular medical check-ups. Early diagnosis through genetic testing can lead to prompt treatment, which significantly improves the prognosis and quality of life. Awareness of these risk factors enables at-risk individuals to take proactive steps in managing their health.
Wilson’s Disease FAQs
Here are the top 10 frequently asked questions about Wilson’s disease, along with detailed answers to help you understand this rare genetic disorder.
- What is Wilson’s disease?
Wilson’s disease is a rare inherited genetic disorder that causes excessive accumulation of copper in the body, particularly in the liver, brain, and eyes. This happens because of a mutation in the ATP7B gene, which impairs the body’s ability to eliminate excess copper. Over time, the buildup of copper can lead to life-threatening organ damage affecting the liver and nervous system if not treated promptly. - What causes Wilson’s disease?
The disease is caused by inheriting defective copies of the ATP7B gene from both parents. This gene is responsible for producing a protein that helps transport excess copper into bile for excretion from the body. Mutations in the ATP7B gene disrupt this process, leading to copper accumulation in vital organs over time. - Who is at risk for Wilson’s disease?
Individuals with a family history of Wilson’s disease are at the highest risk. If both parents carry one defective ATP7B gene, their children have a 25% chance of inheriting two defective genes and developing the disease. Symptoms typically appear between the ages of 5 and 35, but the condition can manifest at any age. - What are the common symptoms of Wilson’s disease?
Symptoms vary depending on the organs affected. Liver-related symptoms include fatigue, weakness, jaundice (yellowing of the skin and eyes), abdominal swelling, and pain. Neurological symptoms involve tremors, muscle stiffness, difficulty with speech or swallowing, poor coordination, and involuntary movements. Psychiatric symptoms may include depression, mood swings, changes in behavior, and cognitive difficulties. A distinctive eye symptom is the presence of Kayser-Fleischer rings, which are golden-brown discolorations around the corneas. - How is Wilson’s disease diagnosed?
Diagnosis involves several tests. Blood tests measure copper levels, ceruloplasmin (a copper-binding protein), and assess liver function. A 24-hour urine copper test evaluates copper excretion in urine over a day. An eye examination using a slit-lamp detects Kayser-Fleischer rings. A liver biopsy examines liver tissue for copper content. Genetic testing identifies mutations in the ATP7B gene to confirm the diagnosis. - How is Wilson’s disease treated?
Treatment aims to reduce copper accumulation. Chelating agents like penicillamine or trientine bind copper, allowing it to be excreted in urine. Zinc therapy with zinc acetate reduces copper absorption from the gastrointestinal tract. A low-copper diet involves avoiding foods high in copper, such as shellfish, nuts, chocolate, mushrooms, and organ meats. In severe cases with extensive liver damage, a liver transplant may be necessary. - Can Wilson’s disease be cured?
While there is no cure for Wilson’s disease, early diagnosis and consistent treatment can effectively manage the condition. Lifelong therapy is usually required to prevent copper buildup. With proper management, individuals can lead normal, healthy lives and prevent serious complications. - Is Wilson’s disease contagious?
No, Wilson’s disease is not contagious. It is a genetic disorder inherited from parents and cannot be transmitted from person to person through contact or other means. - Can Wilson’s disease be prevented?
Since it is a genetic condition, Wilson’s disease cannot be prevented. However, genetic counseling can help at-risk families understand their chances of passing the disease to their children. Early screening and diagnosis are crucial for effective management. - What happens if Wilson’s disease is left untreated?
If left untreated, Wilson’s disease can lead to severe complications, including liver failure due to chronic liver damage and cirrhosis, permanent neurological damage resulting in movement disorders and cognitive impairments, worsening psychiatric disorders, kidney problems affecting renal function, and ultimately death due to severe organ failure. Early detection and treatment are essential to prevent irreversible organ damage and improve the quality of life.
Being informed about Wilson’s disease is vital for early intervention and effective management. If you suspect you or a family member may be at risk, consult a healthcare professional for guidance and appropriate testing.
